CLINICAL TRIAL OVERVIEW
TAZVERIK efficacy and safety were evaluated in 2 open-label, single-arm cohorts (Cohorts 4 and 5) of a multicenter study in patients with histologically confirmed follicular lymphoma after at least 2 prior systemic therapies (n=99). The primary efficacy endpoint was the overall response rate (ORR) and a secondary efficacy endpoint was duration of response (DOR).1,2
WT
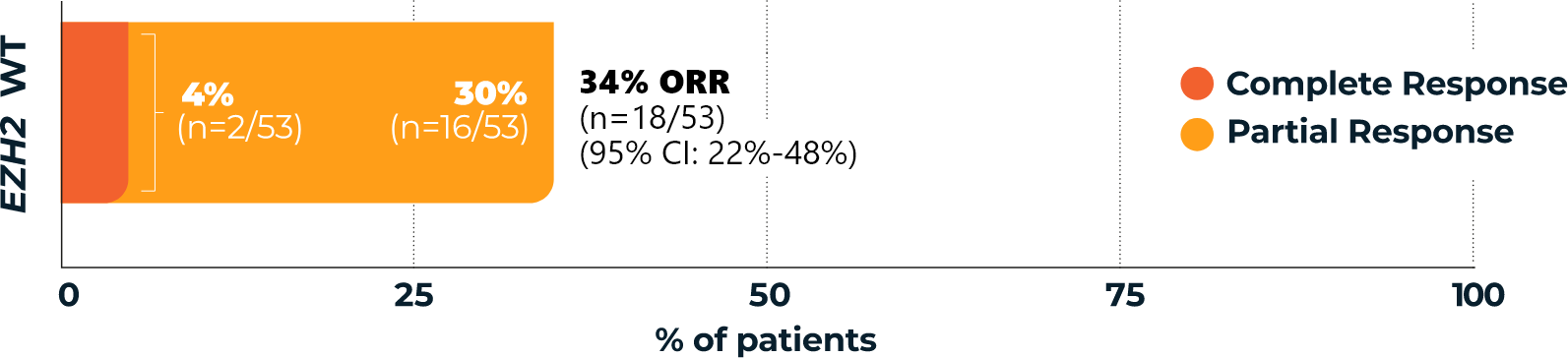
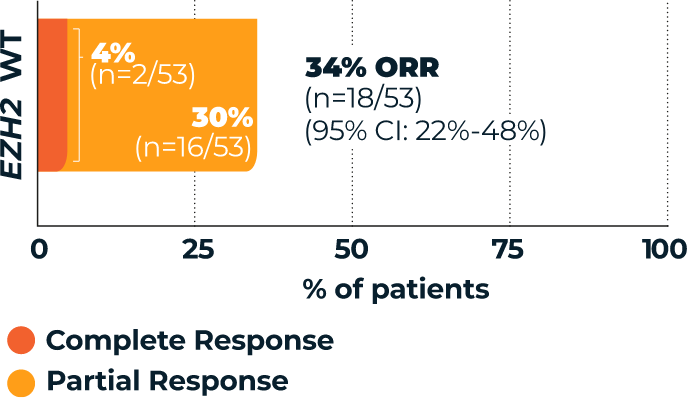
TAZVERIK demonstrated efficacy in EZH2 wild-type patients1
Overall Response Rate
(ORR, primary endpoint)1,3*
Duration of response
(DOR, secondary endpoint)1,3

(range: 1.0-≥22.5 months)
(n=18/53; 95% CI: 5.6-NE)1,2
Of those patients who had a response,3†
- 56% (n=10/18) responded for >6 months
- 39% (n=7/18) responded for >12 months
Median time to overall response for patients with EZH2 WT FL was 3.9 months (range: 1.6 to 16.3)1
- *According to the International Working Group Non-Hodgkin Lymphoma (IWG-NHL) criteria as assessed by Independent Review Committee.1
- †Percentages are based on the intent-to-treat patients within each group who achieved a complete response or partial response.3
MT
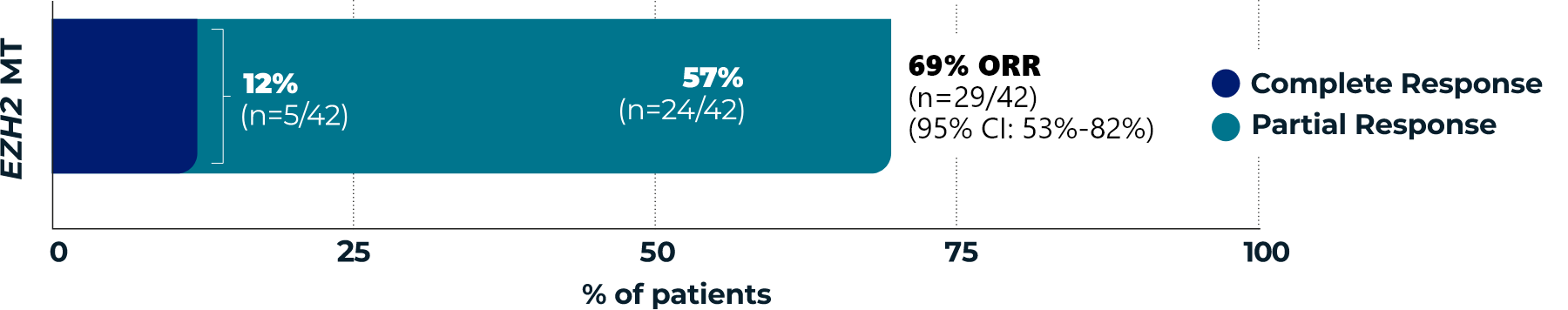
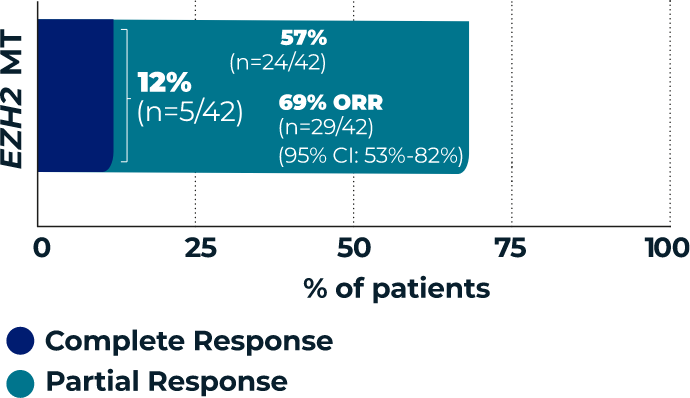
TAZVERIK demonstrated efficacy
in EZH2 mutant-type patients1
Overall Response Rate
(ORR, primary endpoint)1,3*
Duration of response
(DOR, secondary endpoint)1,3

(range: 0.0-≥22.1)
(n=29/42; 95% CI: 7.2-NE)1,2
Of those patients who had a response,3†
- 59% (n=17/29) responded for >6 months
- 21% (n=6/29) responded for >12 months
Median time to overall response for patients with EZH2 MT FL was 3.7 months (range: 1.6 to 10.9)1
- *According to the International Working Group Non-Hodgkin Lymphoma (IWG-NHL) criteria as assessed by Independent Review Committee.1
- †Percentages are based on the intent-to-treat patients within each group who achieved a complete response or partial response.3
EZH2=enhancer of zeste homolog 2; WT=wild type; CI=confidence interval; NE=not evaluable; MT=mutant type.
References: 1. TAZVERIK (tazemetostat) Prescribing Information. Cambridge, MA: Epizyme, Inc., August 2024. 2. Morschhauser F, Tilly H, Chaidos A, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020;21(11):1433-1442. doi:10.1016/S1470-2045(20)30441-1. 3. Data on File.